Teo cơ tủy sống là một rối loạn di truyền trong đó một người không thể kiểm soát chuyển động của cơ bắp của họ, do mất tế bào thần kinh trong tủy sống và não.
Kết quả suy nhược cơ và yếu, khiến khó đứng, đi, kiểm soát chuyển động đầu, và thậm chí, trong một số trường hợp, để thở và nuốt.
Teo cơ tủy sống (SMA) không chỉ là một bệnh mà là một loạt các bệnh. Nhóm lại với nhau, các loại khác nhau của SMA tạo thành nguyên nhân thứ hai dẫn đến bệnh thần kinh cơ.
SMA hiện diện ở 1 trong mỗi 6.000 đến 10.000 ca sinh sống. Đây là chứng rối loạn lặn tự phát tử vong phổ biến thứ hai sau xơ hóa nang.
Nếu SMA ảnh hưởng đến trẻ sơ sinh dưới 2 tuổi, nó thường gây tử vong. Nếu bệnh nhân lớn hơn khi bệnh xuất hiện, họ sẽ có một loại SMA khác nhau và tuổi thọ có thể bình thường.
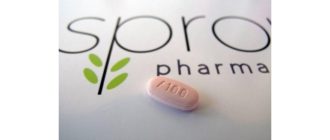
Không có cách điều trị bệnh SMA, nhưng vào tháng 12 năm 2016, loại thuốc đầu tiên được điều trị đã được phê duyệt: Spinraza.
Thông tin nhanh về teo cơ tủy sống:
Dưới đây là một số điểm chính về teo cơ tủy sống (SMA). Chi tiết hơn nằm trong bài viết chính.
- Có nhiều loại SMA khác nhau. Chúng khác nhau về mức độ nghiêm trọng.
- Nó là một tình trạng di truyền.
- Triệu chứng chính là yếu cơ và mất cơ. Trong trường hợp nặng, các vấn đề hô hấp có thể xảy ra.
- Phương pháp điều trị bao gồm các thiết bị hỗ trợ để giúp bệnh nhân hít thở, và một loại thuốc mới, Spinraza.
- SMA không thể được ngăn chặn, nhưng cha mẹ tương lai có thể yêu cầu xét nghiệm di truyền nếu họ có thể là người mang mầm bệnh.
Các loại
Có nhiều loại SMA khác nhau. Chúng khác nhau về mặt khi chúng bắt đầu xuất hiện trong một cá nhân, và trong tuổi thọ mà một người bị rối loạn sẽ có.
SMA loại I

SMA loại I là một tình trạng nghiêm trọng. Trẻ em bị rối loạn này không bao giờ có thể ngồi hoặc đứng. Nó thường gây tử vong trước tuổi 2 năm.
Nó có thể được phát hiện trước khi sinh, vì có thể làm giảm sự chuyển động của thai nhi trong những tháng cuối của thai kỳ. Nếu không, nó sẽ trở nên rõ ràng trong vài tháng đầu đời.
Trẻ sơ sinh mắc bệnh SMA Tôi không bao giờ ngồi hoặc đứng, và chúng thường không sống sót đến tuổi hai năm. SMA type I còn được gọi là bệnh Werdnig-Hoffmann.
SMA loại II
SMA loại II thường xuất hiện trong độ tuổi từ 3 đến 15 tháng. Trẻ sơ sinh có thể học ngồi, nhưng sẽ không bao giờ có thể đứng hoặc đi.
Tuổi thọ phụ thuộc vào việc bệnh nhân có phát triển các vấn đề về hô hấp hay không. Hầu hết những người mắc bệnh SMA loại II sống sót khi trưởng thành.
SMA loại III
Bệnh SMA loại III hoặc bệnh Kugelberg-Welander xuất hiện từ 2 đến 17 tuổi.
Các triệu chứng bao gồm dáng đi bất thường và khó khăn khi chạy, leo bậc thang, hoặc đứng dậy từ ghế. Có thể có một chút rung của ngón tay. Một số người có thể mất khả năng đi bộ, và họ cũng có thể bị vẹo cột sống. Các biến chứng bao gồm béo phì và loãng xương.
Hội chứng Kennedy
Hội chứng Kennedy còn được gọi là chứng teo cơ spinobulbar tiến triển. Hội chứng Kennedy là một tình trạng di truyền, tiến triển chậm thường xuất hiện từ 20 đến 40 tuổi, nhưng nó có thể xuất hiện sau đó.
Phụ nữ mang gen này, nhưng chỉ có một đứa con trai sẽ thừa hưởng sự rối loạn này.
SMA bẩm sinh với arthrogryposis
SMA bẩm sinh với arthrogryposis là một chứng rối loạn hiếm gặp. Những người có tình trạng này sẽ có một co thắt dai dẳng các khớp xương, được gọi là arthrogryposis.
Tình trạng này là hiển nhiên khi sinh. Các tính năng bao gồm co thắt nghiêm trọng, độ cong của cột sống, biến dạng ngực, các vấn đề hô hấp, hàm nhỏ bất thường và mí mắt trên.
Adult SMA
SMA người lớn, hoặc SMA IV, bắt đầu sau 18 tuổi. Những người bị tình trạng này có thể đi lại và họ không có vấn đề gì với việc hít thở hoặc ăn uống.
Các nhà nghiên cứu cũng tìm thấy mối liên hệ giữa SMA và bệnh xơ cứng teo cơ (ALS), thường được gọi là bệnh Lou Gehrig.
Triệu chứng
Các triệu chứng của SMA phụ thuộc vào mức độ nghiêm trọng của nó và tuổi của người đó khi nó bắt đầu. Trẻ sơ sinh mắc bệnh SMA loại I được sinh ra với rất ít cơ bắp, cơ yếu, và các vấn đề về ăn và thở. Với SMA type III, các triệu chứng có thể không xuất hiện cho đến năm thứ hai của cuộc đời.
Trong tất cả các dạng của nó, đặc điểm cơ bản của SMA là yếu cơ, kèm theo teo cơ. Đây là kết quả của việc bảo quản, hoặc mất tín hiệu để ký hợp đồng, được truyền từ tủy sống.
Tín hiệu này thường được truyền từ tế bào thần kinh vận động trong tủy sống đến cơ thông qua sợi thần kinh của động cơ. Trong SMA, hoặc nơron vận động với sợi trục của nó, hoặc chính sợi trục, trở nên không hoạt động. Nó ngừng hoạt động.
Nhiều triệu chứng của SMA liên quan đến các biến chứng thứ cấp của yếu cơ. Đây có thể được thuyên giảm một phần bằng cách điều trị.
Nguyên nhân
SMA xảy ra khi các tế bào thần kinh vận động trong tủy sống và não bộ không hoạt động hoặc ngừng hoạt động, vì những thay đổi di truyền. Các tế bào thần kinh vận động là các tế bào thần kinh điều khiển chuyển động.
Mỗi tế bào người có chứa một phần nhận được hướng dẫn từ các gen, và khi các hướng dẫn chứa một sai lầm, điều này được gọi là xóa. Phần nhận được hướng dẫn thường là một protein.
Trong SMA, các hướng dẫn được trao cho các tế bào thần kinh vận động, hoặc các dây thần kinh điều khiển chuyển động, có chứa một sự xóa gây ra sự thiếu hụt protein. Gen chịu trách nhiệm hướng dẫn các nơron vận động là nơron vận động tồn tại, thường là SMN 1.
Mỗi người có 2 cặp gien cho mỗi chỉ dẫn. Một gen được thừa kế từ mẹ và một từ cha. Một số bệnh sẽ xuất hiện nếu chỉ có một trong các gen di truyền có chứa một lỗi hướng dẫn. Các bệnh khác, chẳng hạn như SMA, sẽ chỉ xuất hiện nếu có một sai lầm trong cả gen của người mẹ và của cha.
Đối với trẻ bị SMA, cả cha lẫn mẹ phải đóng một SMN 1 với hướng dẫn bị lỗi.
Tuy nhiên, ngay cả khi cả hai cha mẹ có gen bị lỗi, đứa trẻ sẽ không phải luôn luôn kế thừa nó. Ngay cả trong số này, cơ hội chỉ là 1 trong 4 cho mỗi thai kỳ mà trẻ sẽ bị SMA. Một trong 40 người lớn là những người mang gen gây bệnh SMA.
Chẩn đoán
Chẩn đoán thường bắt đầu khi cha mẹ hoặc người chăm sóc nhận thấy các triệu chứng của SMA ở trẻ.
Một bác sĩ sẽ thực hiện một lịch sử y tế chi tiết, một lịch sử gia đình, và khám sức khỏe. Họ sẽ kiểm tra xem các cơ có mềm hay không, để kiểm tra phản xạ gân sâu và sự phát triển cơ bắp của cơ lưỡi.
Các xét nghiệm được sử dụng để chẩn đoán SMA bao gồm xét nghiệm máu, sinh thiết cơ, xét nghiệm di truyền, và điện tâm đồ tiềm năng (EMG). EMG được sử dụng để đánh giá sức khỏe của cơ bắp và tế bào thần kinh, hoặc tế bào thần kinh vận động, điều khiển chúng. Chọc nước ối hoặc lấy mẫu lông nhung chorionic có thể đánh giá thai nhi trong thời kỳ mang thai.
Điều trị
Có, như được nêu ra, không có chữa bệnh cho SMA, và không có cách nào để ngăn chặn nó, vì nó là một condtion kế thừa. Tuy nhiên, điều trị có thể giúp mọi người sống cuộc sống đầy đủ hơn.
Spinraza
Vào tháng 12 năm 2016, Cơ quan Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) đã chấp thuận một loại thuốc, nusinersen (Spinraza) để điều trị SMA. Đây là loại thuốc đầu tiên được chấp thuận cho tình trạng này.
Nó được tiêm, ba liều đầu tiên trong khoảng thời gian 14 ngày, liều thứ tư sau 30 ngày, và sau đó cứ 4 tháng một lần.
Spinraza nhắm vào các khiếm khuyết cơ bản trong SMA, vì vậy nó có thể giúp trì hoãn, ngăn ngừa hoặc thậm chí đảo ngược các triệu chứng. Các tác dụng phụ thường gặp bao gồm nguy cơ nhiễm trùng đường hô hấp và táo bón cao hơn. Cũng có thể có nguy cơ chảy máu và các vấn đề về thận.
Thiết bị hỗ trợ
Công nghệ hỗ trợ như máy thở, xe lăn điện, và truy cập được sửa đổi cho máy tính là cho phép những người bị SMA sống lâu hơn, năng động hơn và tham gia cộng đồng.
Thông gió đặc biệt quan trọng. Mức độ nghiêm trọng của điểm yếu của cá nhân trực tiếp ảnh hưởng đến quá trình của bệnh. Trẻ sơ sinh bị SMA nặng có thể bị bệnh hô hấp, vì các cơ hỗ trợ hơi thở yếu.
Trẻ em có dạng SMA nhẹ hơn có thể mong đợi tuổi thọ dài hơn, mặc dù chúng có thể cần được hỗ trợ y tế rộng rãi.
Sinh học phân tử đã cải thiện sự hiểu biết của chúng ta về SMA. Nhiều phương pháp thí nghiệm đang được thử nghiệm, bao gồm thay thế gen, thay thế tế bào gốc của tế bào thần kinh vận động và các liệu pháp để tăng sự biểu hiện của gen SMN 2.
SMA là di truyền, và không có cách nào để ngăn chặn nó.
Phụ huynh có tiền sử gia đình mắc bệnh SMA được khuyến khích tìm tư vấn di truyền trước khi bắt đầu một gia đình.